La fibrosi cistica è una malattia genetica che colpisce i polmoni e altri sistemi del corpo. Attualmente non esiste cura, ma grazie ai progressi nella diagnostica e nelle cure, oggi chi soffre di fibrosi cistica ha un tasso di sopravvivenza e una qualità della vita migliore rispetto a chi ne soffriva nei decenni precedenti.
La fibrosi cistica è una malattia genetica ereditaria (che si eredita dai genitori proprio come il colore degli occhi o la forma del naso) e colpisce i bambini e i giovani adulti. Nel mondo ci sono circa 70.000–100.000 casi di fibrosi cistica e ogni anno se ne aggiungono altri 1.000.1,2 La fibrosi cistica è più comune nei bambini di etnia caucasica e colpisce un bambino ogni 2500–3000 neonati. L’incidenza della fibrosi cistica è meno comune in altri gruppi etnici.3
Chi soffre di fibrosi cistica ha una proteina difettosa che regola il movimento del sale e dell’acqua all’interno e all’esterno delle cellule. Questo difetto è causato dalla mutazione o alterazione di un gene, il gene regolatore della conduttanza transmembrana della fibrosi cistica (CFTR).1
La fibrosi cistica agisce sulle cellule che producono muco, sudore e succhi digestivi. Nella maggior parte dei casi la fibrosi cistica colpisce i polmoni e il sistema digestivo, ma può attaccare anche i seni paranasali, il fegato, il pancreas e gli organi riproduttivi (Figura 1). In condizioni normali, questi organi producono ed eliminano i fluidi, noti anche come secrezioni, e agiscono da lubrificanti. Tuttavia, in chi soffre di fibrosi cistica queste secrezioni diventano appiccicose e dense e bloccano i dotti escretori a livello di vari tessuti, in particolare nei polmoni e nel pancreas.4,5
Figura 1 - Parti del corpo colpite da fibrosi cistica.4
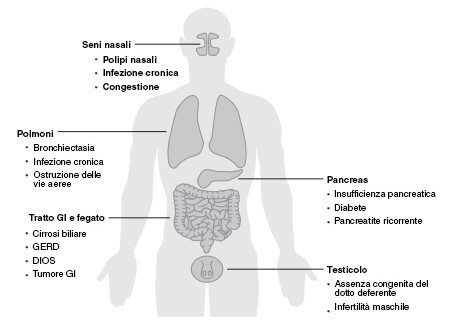
DIOS: sindrome da ostruzione dell’intestino distale (ovvero blocco della parte distale dell’intestino tenue); GERD: malattia da reflusso gastroesofageo (un disturbo digestivo che si verifica quando i succhi acidi dello stomaco, il cibo o i liquidi risalgono dallo stomaco all’esofago, causando acidità); GI: sistema gastrointestinale. Bronchiectasia: una patologia che danneggia le vie aeree dei polmoni, rendendo difficile l’eliminazione del muco. Polipi nasali: escrescenze benigne e indolori sulla membrana del naso o dei seni paranasali. Assenza congenita del dotto deferente: assenza del dotto deferente dalla nascita. Il dotto deferente fa parte del sistema riproduttivo maschile e trasporta lo sperma dai testicoli al pene.
Chi è colpito da fibrosi cistica?
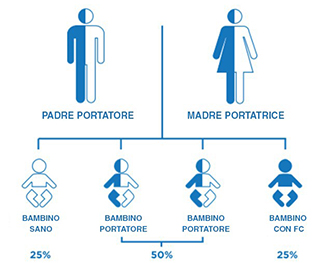
La fibrosi cistica è una malattia genetica che si verifica nei bambini con due geni difettosi, ereditati uno da ogni genitore (Figura 2). Quando due individui con un difetto genetico responsabile della fibrosi cistica hanno un figlio, esistono6:
- il 25% di probabilità che il bambino nasca con la fibrosi cistica
- il 50% di probabilità che il bambino sarà portatore di un gene difettoso responsabile della fibrosi cistica
- il 25% di probabilità che il bambino non sarà portatore né avrà la fibrosi cistica
Se la diagnosi di fibrosi cistica è confermata, gli altri membri della famiglia, compresi fratelli e sorelle, dovranno sottoporsi a un controllo. Inoltre, si consiglia di controllare anche i parenti adulti per verificare il loro stato di portatori in modo che possano sottoporsi a esami di screening prenatale (prima della nascita, durante la gravidanza).7
Figura 2 - Chi è colpito da fibrosi cistica?6
I segni e sintomi della fibrosi cistica variano a seconda della gravità della malattia. Chi soffre di fibrosi cistica ha il sudore salato. Altri segni e sintomi della fibrosi cistica interessano il sistema respiratorio e digestivo. Tuttavia, gli adulti a cui è stata diagnosticata la fibrosi cistica presentano sintomi strani, come episodi ricorrenti di infiammazione al pancreas (pancreatite), polmonite ricorrente e infertilità.5
- Segni e sintomi respiratori: tosse ricorrente con espettorato; affanno; respiro corto; infezioni polmonari ripetute e cavità nasali infiammati o naso chiuso5,6
- Segni e sintomi digestivi: feci untuose e dall’odore fetido; scarso aumento di peso e della crescita; blocco intestinale, in particolare nei neonati (ileo da meconio) e grave stipsi5–7
Steatorrea e scarso aumento di peso sono attribuiti all’insufficienza pancreatica esocrina (IPE). La fibrosi cistica è una delle principali cause dell’IPE; circa l’80%–90% delle persone con fibrosi cistica sviluppa IPE.8 Nella maggior parte delle persone con fibrosi cistica l’IPE viene diagnosticata nel primo anno di vita. L’insufficienza pancreatica esocrina è considerata la causa principale del malassorbimento e della malnutrizione nei pazienti con fibrosi cistica. Quindi, l’obiettivo principale è curare la maldigestione e il malassorbimento delle sostanze nutritive ricorrendo alla terapia sostitutiva con enzimi pancreatici in base alla gravità della steatorrea.9
La fibrosi cistica è una malattia limitante. Tuttavia, oggi, grazie ai progressi nella diagnostica e nella terapia, il tasso di sopravvivenza è migliorato. Ora chi soffre di fibrosi cistica può vivere per più di 40 anni, rispetto ai soli otto anni nel 1970. Infatti, 40 anni fa solo il 10% dei pazienti con fibrosi cistica superava i 18 anni di età, mentre ora oltre il 50% di loro potrebbe diventare maggiorenne.3,4
Nonostante i pazienti con fibrosi cistica richiedano cure giornaliere e a volte affrontino grandi difficoltà nella vita quotidiana, di solito sono in grado di andare a scuola e lavorare e spesso la loro qualità della vita è migliore rispetto a chi soffriva di fibrosi cistica nei decenni precedenti.5
-
Bibliografia:
- Schneider-Futschik EK. Beyond cystic fibrosis transmembrane conductance regulator therapy: A perspective on gene therapy and small molecule treatment for cystic fibrosis. Gene Therapy. 2019;26:354–362.
- Rafeeq MM, Murad HAS. Cystic fibrosis: Current therapeutic targets and future approaches. J Transl Med. 2017;15:84–92.
- Australasian guidelines for the management of pancreatic exocrine insufficiency. 2015. Available at: http://pancreas.org.au/wp-content/uploads/2016/01/APC-GUIDELINES-2015.pdf. Accessed on: 2 April 2019.
- Rey MM, Bonk MP, Hadjiliadis D. Cystic fibrosis: Emerging understanding and therapies. Annu Rev Med. 2019;70:197–210.
- Cystic fibrosis. Available at: https://www.mayoclinic.org/diseases-conditions/cystic-fibrosis/symptoms-causes/syc-20353700. Accessed on: 16 November 2019.
- What is cystic fibrosis? Available at: https://www.cysticfibrosis.ca/about-cf/what-is-cystic-fibrosis. Accessed on: 16 November 2019.
- Davies JC, Alton EWFW, Bush A. Cystic fibrosis. BMJ. 2007;335:1255–1259.
- Kashirskaya NY, Kapranov NI, Sander-Struckmeier S, et al. Safety and efficacy of Creon® Micro in children with exocrine pancreatic insufficiency due to cystic fibrosis. J Cyst Fibros. 2015;14:275–281.

Cos’è l’IPE?
Se il pancreas non funziona correttamente per un periodo di tempo prolungato, la secrezione o il funzionamento degli enzimi pancreatici potrebbe risultare insufficiente. Questa condizione è chiamata insufficienza pancreatica esocrina (IPE).1

Cura dell’IPE
Il vostro medico farà una valutazione nutrizionale e se risulterà uno stato nutrizionale compromesso, potrà prescrivere degli integratori nutrizionali e suggerirà altre modifiche allo stile di vita.